PO48 | Thrombotic risk evaluation and thrombophilia testing in β-thalassemia patients: a retrospective cohort analysis from a reference regional center
G.M. Camarda1,2*, S. Raso1*, R. Di Maggio1, M. Vinciguerra3, A. Giangreco1, A. Inzerillo1, A. Maggio, M. Napolitano1,2 | *These authors contributed equally to this abstract; 1Hematology and Rare Diseases Unit, Azienda Ospedaliera Ospedali Riuniti Villa Sofia-Cervello, Palermo; 2Division of Hematology Department of Health Promotion, Mother and Child Care, Internal Medicine and Medical Specialties “ProMISE”, Palermo; 3UOSD Molecular Diagnosis of Rare Blood Diseases, Azienda Ospedaliera, Ospedali Riuniti Villa Sofia, Cervello, Palermo, Italy
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Authors
Background: β-thalassemia (βT) is a group of inherited hemoglobin synthesis disorders, characterized by defective β-chain synthesis. Clinical complications related to hemostasis, including pulmonary hypertension, venous thromboembolism (VTE), and ischemic arterial events, occur in βT with a prevalence ranging from 1.1% to 5.3%. The exact cause of the hypercoagulable state in βT patients remains unclear, though several factors have been advocated. Thrombophilia might contribute to VTE in βT, but studies do not show an increased frequency of inherited thrombophilia conditions in βT patients. However, more well-structured studies are needed to reach definitive conclusions.
Aims: This retrospective analysis aimed to investigate the clinical context and indications for thrombophilia screening in βT patients.
Methods: A retrospective analysis was conducted on hospital records and patient charts of βT patients managed at our Reference Regional Center over the past five years, focusing on the clinical context and indications for thrombophilia screening.
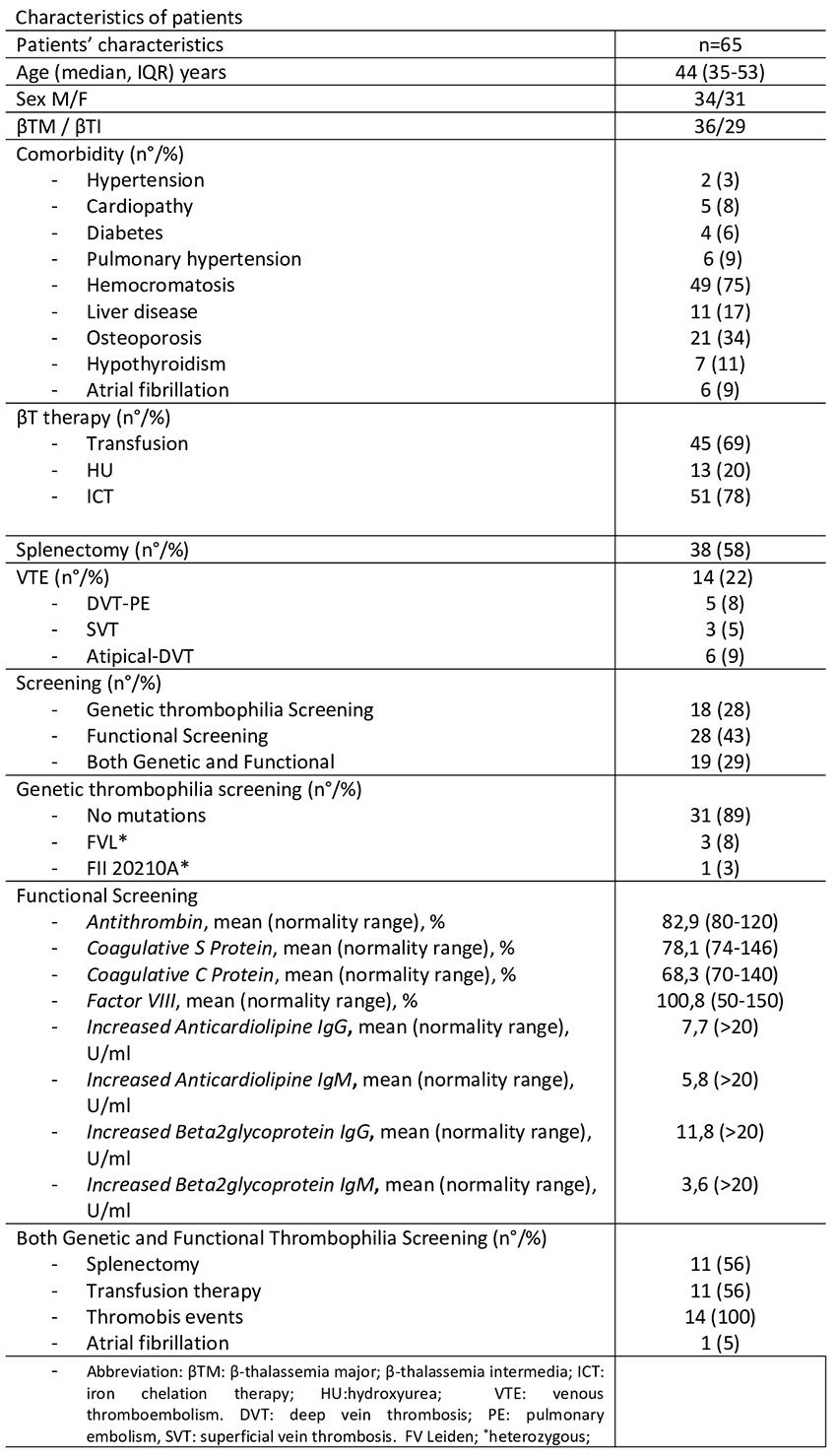
Results: Sixty-five βT patients underwent thrombophilia screening over five years. Nineteen had complete screening (including both functional and genetic testing), while 18/65 and 28/65 patients had partial genetic and functional screening, respectively (Table 1). Complete thrombophilia screening was mostly performed in patients undergoing splenectomy (56%) and with a personal history of VTE (100%) to assess thrombotic risk and guide anticoagulant therapy in the absence of specific guidelines. Thrombotic events were the second most common complication after hemochromatosis. None of the patients were homozygous for FII (G20210A) or FV Leiden gene mutations. One patient (3%) was heterozygous for FII (G20210A) and three (8%) for FV Leiden. Functional screening revealed elevated anticardiolipin antibodies (aCL) and anti-β2-glycoprotein I antibodies (anti-β2GPI) in less than 12% of patients, with lupus anticoagulant (LAC) absent.
Conclusions: The lower prevalence of antiphospholipid antibodies (aPLs) in our cohort, compared to existing literature, highlights the variability of aPL prevalence in βT patients and the complexities in interpreting these findings. Moreover, the low prevalence of common genetic mutations and antiphospholipid antibodies suggests that other factors may contribute more significantly to thrombotic risk in βT patients. Further research is needed to explore alternative genetic and acquired thrombophilic conditions in this population. Given the known morbidity caused by thrombotic events in βT, our data underscore the importance of personalized risk assessment in clinical practice.
Downloads
Citations




How to Cite

This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.