PO65 | Utilization of emicizumab in acquired hemophilia A: a case report
C. Caputo, P. Conca, I.L. Calcaterra, E. Cimino, M. Romeo, M. Aversano, E. Franco, C. De Luca, G. D’errico, L. Jr Valletta, M. Di Minno, A. Tufano | Department of Clinical Medicine and Surgery, Federico II University, Naples, Italy
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Authors
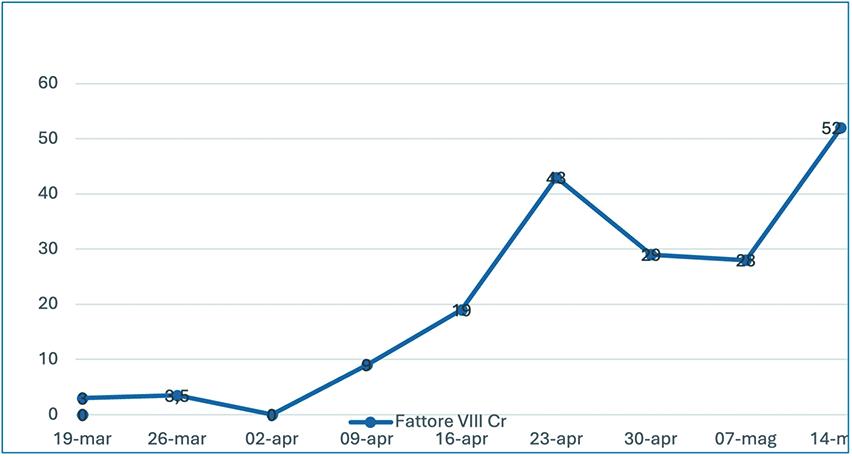
Background: Acquired hemophilia A (AHA) is a severe bleeding disorder caused by autoantibodies against coagulation Factor VIII (FVIII). Treatment consists of bleeding control with bypassing agents and immunosuppressive therapy (IST). Emicizumab, a bispecific antibody that mimics the function of activated FVIII irrespective of the presence of neutralizing antibodies, demonstrated to prevent bleeds and to allow to postpone or individualize the immunosuppression. Case Report: An 81-year-old female patient presented with spontaneous mucocutaneous bleedings, including diffuse hematomas and severe posterior epistaxis, in the absence of a known coagulopathy. These symptoms were associated with acute severe anemia, requiring multiple red blood cells (RBC) transfusions. Her medical history included previous diagnosis of breast cancer in follow-up, widespread atherosclerosis, arterial hypertension, and severe osteoporosis. Baseline coagulation studies revealed an abnormally prolonged activated partial thromboplastin time (aPTT ratio of 2.7), with normal PT/INR. A mixing test was performed with no correction. Acquired hemophilia was suspected with subsequent clot-based FVIII activity of 3% and FVIII inhibitor of 8.7 BU/ mL, findings that prompted the initiation of recombinant Factor VIIa (rFVIIa; 90 mcg/kg every 6 h), and tranexamic acid, as well as IST with prednisone (80 mg, daily). No significant clinical or laboratory improvement was observed after two weeks. Treatment with rituximab (375 mg/m² weekly for four doses) was then introduced and steroid treatment was progressively reduced. To prevent bleeding episodes in the presence of high thromboembolic risk, rFVIIa was stopped and the treatment with Emicizumab was started with the following therapy schedule: 6 mg/kg on day 1, 3 mg/kg on day 2; maintenance: 3 mg/kg after a further 7 days and every subsequent 14 days until chromogenic FVIII of 20% was reached. A progressive and significant reduction in inhibitor levels was observed, confirming the efficacy of anti-CD20 therapy with rituximab. Furthermore, the early use of emicizumab contributed to effective hemostatic control, as evidenced by stabilization of hemoglobin levels, resolution of cutaneous hemorrhagic manifestations, and the absence of new spontaneous bleeding episodes throughout the follow-up period. No further substitution of coagulation factors was needed. Conclusions: These findings underscore the therapeutic value of Emicizumab in patients with AHA and complex clinical profiles, demonstrating the efficacy in the reduction in bleeding risk throughout the course of treatment.
Downloads
Citations




How to Cite

This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.